Basic cellular reactions of the
immature human brain
Waney Squier
Chapter Contents
- Introduction
- The postmortem examination
- Cellular reactions of the immature human
- Hemorrhages
- White matter damage
- Gray matter damage
- Malformations
- Infections
- Summary
- References
Introduction
An understanding of basic neuropathological processes is fundamental to the interpretation of brain scans. In this chapter basic cellular reactions to injury of the developing human brain are described, with special reference to those processes which may be identified on scans.
Those patterns of injury most characteristic of, and most frequently seen in, the developing brain are described. They have to be interpreted in the light of the changing anatomy of the developing brain. For a comprehensive atlas of the anatomy of the immature brain the reader is referred to Feess-Higgins and Larroche8.
This chapter deals with reactive processes to a variety of insults. Although hypoxic–ischemic injury (HII) may be a very common cause of damage to the developing brain it is clear that many other insults play a part in prenatal brain damage3. These include maternal and fetal infections, iodine deficiency and genetic, including metabolic, diseases.
< prev | top | contents | next >
The postmortem examination
WHY PERFORM A POSTMORTEM EXAMINATION?
- To establish a cause of death
- To diagnose disease
- To assess therapy
- To audit new imaging techniques
- To further understanding of normal and abnormal brain development
- Informed consent for brain retention is required.
Until the last two decades the importance of postmortem examination of the fetus and neonate has been underestimated.
The primary purpose of postmortem examination is to establish a cause of death and to diagnose disease. It can reveal pathology too subtle to be identified by clinical investigations or imaging.
Full examination of all organs, including the placenta, will reveal generalized disease processes which may have detrimental effects on brain development. Examination of the placenta is of particular importance; recent studies show an association of placental pathology, particularly infection, with developmental brain damage11, 12.
Complete postmortem examination requires detailed macroscopic and microscopic examination of the brain. This involves retention of the brain with the informed consent of the parents.
The brain has to be fixed before it can be adequately examined. In small fetuses the brain is very soft and collapses readily on handling and is best preserved by fixation in situ. This not only ensures optimal brain fixation but also preserves the relationships of the intracranial structures (Fig. 5.1). In older fetuses (after 30 weeks) and neonates the brain can be removed at postmortem and then fixed for at least 2–3 weeks before macroscopic examination. Blocks are taken and embedded in wax prior to cutting sections and staining for microscopic study.

Fig. 5.1 Fetal brain (20 weeks) fixed in situ. The posterior fossa has been opened from behind. The anatomy of the hindbrain and its position within the skull is readily seen. Malformations or displacement of hindbrain structures are readily recognized if the brain is examined in this way.
The most widely used stain in light microscopy is hematoxylin and eosin (H&E). Luxol fast blue and cresyl violet (LBCV), another standard neuropathological method, stains nuclei, RNA and myelin. This stain is less useful in the fetal brain where myelin is sparse.
Immunocytochemical methods employ labelled antibodies to detect specific tissue antigens. These are currently widely used to identify specific cell types and their products.
ARTIFACTS
Human tissue is extremely subject to artifactual change. This is because the brain undergoes postmortem autolysis or maceration following death in utero or if there is a delay between death and postmortem.
Commonly encountered artifacts include tissue vacuolation, which occurs in macerated brains and is readily confused with edema. Maceration causes cells to break up and nuclei to fragment; these fragments are small and irregular and readily distinguished from karyorrhexis. In many immature brains large areas of ependyma may be shed. This must be distinguished from pathological ependymal loss with associated gliosis.
< prev | top | contents | next >
Cellular reactions of the human brain
- Edema (1h)
- Cell death: necrosis: apoptosis (1–12h)
- Gliosis (from 3 days)
- Phagocytosis (from 3 days)
- Capillary proliferation (3–8 days)
- Mineralization (from 1 week)
- Ependymal reaction (2–3 days).
Following injury to the developing brain a series of cellular reactions is set in action, the speed and intensity of these reactions being dependent on the age at the time of injury. Histological examination of the brain in the weeks following injury will allow precise timing, based on the stage of the reactive processes. Some of these cellular reactions leave scars in the brain which persist and are detectable on scans in later life.
EDEMA
The immature brain swells rapidly, within an hour of severe injury. Edema is due to swelling of cell bodies, particularly astrocytes and to accumulation of fluid in the interstitial spaces following leakage through damaged capillary endothelium. Brain swelling causes compression of the lateral ventricles and the cortical sulci. Owing to the ample space around the immature brain and unfused skull sutures herniation is rare in infants.
CELL DEATH
Two main forms of cell death are recognized (Fig. 5.2). The passive form of death is necrosis. The cell membrane loses its integrity, calcium floods into the cell and the cytoplasm becomes bright pink in H&E stains. The nuclear membrane breaks down with lysis of nuclear chromatin. Necrosis is seen within hours of insult. Apoptosis is an active form of cell death mediated by a cascade of intracellular enzyme reactions terminating in breakdown of nuclear DNA. The histological appearances are distinctive. Within hours of injury the nucleus becomes shrunken and rounded and is intensely, uniformly basophilic, appearing deep blue in H&E stains. This appearance is known as pyknosis. Subsequently, the nucleus breaks up into a number of rounded fragments or apoptotic bodies. This appearance is termed karyorrhexis. Apoptosis is the mechanism of programmed cell death by which cells produced in excess during normal brain development are removed.
GLIOSIS
Astrocytes (the cells responsible for reactive gliosis) have been demonstrated in human brain sections by immunocytochemistry at 15 weeks’ gestation18. A vigorous glial response can occur as early as 18 weeks’ gestation (Fig. 5.3).
Reactive astrocytes are identified within 3 days of an insult; in the early stages they have ample cytoplasm and few processes; dense fibrillary gliosis takes weeks to appear.
PHAGOYCTOSIS
Macrophages are cells which ingest and remove debris from areas of tissue injury and contribute to cyst formation. They develop from microglia, the intrinsic macrophage population of the brain. The earliest reactive changes are seen within hours of injury but mature macrophages with large amounts of cytoplasm containing ingested debris are not seen until several days after injury (Fig. 5.4). They may persist for months or years.
Following hemorrhage, macrophages begin to ingest red cells within 3 or 4 days (Fig. 5.5). Red cells are identified within the cytoplasm where they are degraded and change from red to brown in color. Iron pigment may be demonstrated within macrophages months and years after a bleed (see Chapter 9).

Fig. 5.2 Cell death. A large neuron is dying by necrosis (arrow). The cytoplasm stains deeply with eosin, the nucleus has lost all structure and is undergoing lysis. Several adjacent cells show the features of apoptosis with shrunken, rounded and intensely staining nuclei. (small arrows) (H&E x 450.)

Fig. 5.3 Gliosis. Deep white matter adjacent to an area of trauma in a fetus of 18 weeks. There are many reactive astrocytes, intensely stained for glial fibrillary acidic protein (GFAP). (GFAP x 200.)
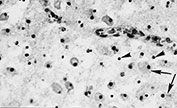

CAPILLARY PROLIFERATION
Capillary endothelium is extremely sensitive to hypoxic–ischemic injury. In the first days after injury the cells become thicker with rounded nuclei; after 5–8 days new capillaries proliferate. Capillary proliferation in the ischemic cerebral cortex is responsible for the MR imaging appearance of ‘cortical highlighting’ (Fig. 5.6) (see Chapter 6).
")
")
")
")
Fig. 5.6 Cortical highlighting. (a) T1 weighted (SE 860/20) MR scan of 2-week-old infant with stage II hypoxic–ischemic encephalopathy (HIE) showing areas of abnormal high signal intensity within the cortex (arrow). (b) CT scan of a 4-month-old child 9 days after trauma and cardiac arrest. The cortex is bright, particularly in the depths of sulci (arrow). (c) Histology of a corresponding area of the cortex shows tissue necrosis with increased cellularity due to gliosis and prominent capillaries, particularly in the depths of sulci. The increased vascularity causes cortical highlighting in scans. (H&E x 25.) (d) At higher power branching, proliferating capillaries are seen in necrotic cortex (arrow). (H&E x 45.)
MINERALIZATION
Mineral deposition occurs readily in damaged areas of the fetal brain (Fig. 5.7). It is seen within 1 week of injury and appears in H&E stained sections as deep blue granules in cell bodies and on nerve processes. It persists for many years. Mineral deposition is responsible for the chalky white color of lesions of periventricular leukomalacia when seen by the naked eye and is characteristic of viral infections when it is widely distributed in areas of damage throughout the brain. Mineralization of the deep cerebral nuclei, especially the thalamus, is seen following old ischemic injury.
")
")
Fig. 5.7 Mineralization. (a) Small cysts in the deep white matter in periventricular leukomalacia (arrows). The edge is chalky white due to mineralization. (b) Thalamus in an infant surviving several years after severe asphyxia. There are several calcified nerve cells (arrow). (H&E x 450.)
EPENDYMAL REACTION
The ependyma is a single layer of epithelium which lines all the cerebrospinal fluid (CSF) pathways of the brain. Damage to the ependyma causes focal loss and reactive proliferation of the underlying glial cells. This response causes narrowing of CSF pathways. Abortive regeneration of ependyma forms rosettes buried in the reactive glial tissue (Fig. 5.8).
")
")
Fig. 5.8 Ependymal reaction. (a) Damaged ependyma in the fourth ventricle. The surface epithelium has been shed in several places (arrows). There is fresh blood in the lumen. (H&E x 120.) (b) Higher power picture shows gliotic tissue bulging through ependymal defects. There are fragments of ependyma in this gliotic tissue (arrows). (H&E x 200.)
< prev | top | contents | next >
Hemorrhages (see also Chapters 9 and 13
- Subdural – term infants – trauma
- Subarachnoid – preterm and term – ischemia
- Parenchymal – rare, severe – multiple etiologies
- Periventricular/intraventricular – sick preterm infants –
association with parenchymal hemorrhagic infarction.
SUBDURAL HAEMORRHAGE
Hemorrhage into the subdural space typically occurs in term infants and is usually the result of trauma. It results from tearing of superficial veins or sinuses on the surface of the brain. Blood clot in the subdural space gradually liquifies from the center. The edges of the clot are invaded by fibroblasts and capillaries and by 10 days the clot is reduced to a brown vascular membrane. The delicate new capillaries of the membrane may rebleed after even minor trauma.
Macrophages ingest and break down red cells and may remain for months. This explains the low signal intensity from hemosiderin on T2 weighted images which persists for months from a perinatal hemorrhagic lesion (see Chapter 9). The altered blood pigment stains the surface of the brain bright orange or yellow.
SUBARACHNOID HEMORRHAGE
Primary subarachnoid hemorrhage is commonly encountered in premature infants at postmortem but rarely identified with imaging. It is usually mild. It forms a thin, diffuse film over the brain surface. Secondary subarachnoid hemorrhage results from subarachnoid spread of intraventricular blood and forms large collections in the basal subarachnoid spaces. Subarachnoid hemorrhage in the term infant is usually related to severe hypoxia–ischemia or to trauma and may be extensive.
PARENCHYMAL HEMORRHAGE
This is uncommon and may be due to a variety of causes including severe trauma, infections, vasculitis or rupture of a vascular malformation. Parenchymal hemorrhage arising in utero may be due to a clotting disorder such as alloimmune thrombocytopenia.
PERIVENTRICULAR GERMINAL MATRIX/ INTRAVENTRICULAR HEMORRHAGE
Periventricular and intraventricular bleeding is characteristic of the sick premature infant and usually occurs within the first 12–72h of life, but is also observed in a proportion of stillborn infants.
The germinal matrix is the usual source of bleeding. This consists of a dense mass of proliferating cells abundantly supplied with thin-walled capillaries. By 36 weeks’ gestation the matrix involutes and periventricular hemorrhage is less common. A small proportion of intraventricular bleeds originate in the choroid plexus, usually in term infants (see Chapter 9). Autoregulation of the cerebral circulation is disturbed in the sick premature infant17 and the capillary network then becomes susceptible to sudden fluctuations in systemic blood pressure.
GERMINAL MATRIX HEMORRHAGE

Fig. 5.9 Periventricular–intraventricular hemorrhage. Coronal slice of fixed fetal brain, 24 weeks’ gestation. There are bilateral germinal matrix hemorrhages (arrows) with intraventicular extension. On the right side a large area of hemorrhagic parenchymal infarction fans out from the germinal matrix hemorrhage (arrow).
Periventricular hemorrhages may be small and confined to the matrix or may rupture through the ependyma into the lateral ventricles (Fig. 5.9).
Matrix hemorrhages are often multiple and bilateral. They tend to occur over the body of the caudate nucleus in very small infants and more anteriorly, at the level of the foramen of Monroe, in infants over 28 weeks.
Localized masses of blood clot undergo evolution over the days and weeks after they occur with central lysis of the clot and invasion of the periphery by macrophages in the first 2–3 days. After prolonged survival the only residual findings may be granules of iron pigment, sometimes within macrophages or persistence of a smooth-walled subependymal cyst at the site of the original bleed7. Whilst old hemorrhage is easily identified on T2 weighted MR images, subependymal cysts are more difficult to detect.
Alterations in the windowing of the MR images may increase the detection of these cysts, which are usually clearly visible on cranial ultrasound.
INTRAVENTRICULAR HEMORRHAGE
Blood rupturing into the lateral ventricles may be confined to one side or, if more extensive, will spread throughout the ventricular system, through the narrow aqueduct of Sylvius in the mid-brain and into the fourth ventricle. From here blood escapes into the subarachnoid cisterns beneath the brain and cerebellum and surrounds the brain stem. Eventually, blood is washed over the surface of the brain in the CSF and reabsorbed in the arachnoid villi of the sagittal sinus.
Blood in the CSF prompts a brisk reaction in the ependyma and underlying tissues (Fig. 5.8). The ependyma is shed and underlying glial cells proliferate and form masses which protrude into the lumen. Proliferating ependymal cells form rosettes within these gliotic masses which may also contain pigment-bearing macrophages. Narrow parts of the CSF pathways, including the mid-brain aqueduct and the exit foramina of the fourth ventricle, may be obstructed by this reaction and impede the flow of CSF leading to hydrocephalus. When the mid-brain aqueduct is narrowed the lateral and third ventricles dilate. If the exit foramina of the fourth ventricle are obstructed then the lateral, third and fourth ventricles dilate. If blood causes reactive gliosis in the arachnoid villi then resorption of CSF is impaired and there is dilatation of both the intra- and extracerebral CSF spaces causing communicating hydrocephalus.
PARENCHYMAL HEMORRHAGIC INFARCTION
The most serious consequence of germinal matrix/intraventricular hemorrhage is parenchymal hemorrhagic infarction which carries a much more serious prognosis than germinal matrix hemorrhage alone. Parenchymal hemorrhagic infarction occurs in the white matter adjacent to a matrix bleed. The fan-shaped lesion consists of clusters of perivenous hemorrhages extending along veins draining the centrum semiovale (Figs Fig. 5.5 and Fig. 5.9). The parenchyma is infarcted and, in infants who survive, it is gradually reabsorbed leading to unilateral dilatation of the lateral ventricle (see Figs 9.28f and 9.29a). This distinguishes the lesion from periventricular leukomalacia, which more commonly leads to symmetrical posterior dilatation of the lateral ventricles, which are irregular and ‘squared off’ in outline (see Chapter 8)21. Occasionally, particularly in association with infection, the two conditions appear to co-exist (see Figs 10.28g,h).
< prev | top | contents | next >
White matter damage
- Multiple etiologies – perfusion failure – hypoxia – infection – inflammation – metabolic disease.
- Periventricular leukomalacia (PVL) – diffuse with white matter atrophy and gliosis (telencephalic leukoencephalopathy). Focal infarcts of classical PVL. Typically seen between 28 and 36 weeks but can also arise at term.
- Multicystic leukomalacia – multiple cysts, periventricular or subcortical. Usually associated with abrupt severe interference in blood supply between 30 and 44 weeks. May be mimicked by herpes encephalitis.
- Posterior limb of the internal capsule.
The white matter is particularly vulnerable to damage between 28 and 36 weeks’ gestation. This has usually been ascribed to ischemia due to a tenuous vascular supply but recent studies have shown that the deep white matter has an extensive vascular network13. At this stage of development the white matter is undergoing active myelination and the immature oligodendrocytes responsible for myelin synthesis are actively migrating into the area. The vulnerability of those cells to free radical damage may explain white matter damage1.
Although hypoxia–ischemia may be a common cause of white matter damage it is becoming increasingly apparent that many other conditions are associated with prenatal cerebral damage3 Maternal infections have been implicated for some years10 and recent studies have further supported the role of maternal or placental infection with white matter lesions, possibly mediated by cytokines5, 6, 23 .
Prenatal white matter damage takes several forms which are related both to the age at insult and the nature of the insult.
PERIVENTRICULAR LEUKOMALACIA
This term encompasses a spectrum of white matter damage, from mild but diffuse injury (sometimes termed telencephalic encephalopathy) to focal areas of cystic infarction. It is a pathological term and its use to describe a variety of imaging appearances is being actively discouraged.
In diffuse leukomalacia the white matter shows only subtle alteration on naked eye examination. The blood vessels are prominent and occasionally flanked by small yellow streaks or patches. Histology shows diffuse infiltration with plump reactive astrocytes and macrophages. Capillaries have thickened endothelium in the early stages and later proliferate20. In the chronic stage the white matter is reduced in volume and shows dense fibrillary gliosis. This pattern of injury is probably the result of mild chronic insult (Fig. 5.10).
In more severe leukomalacia focal infarcts appear as white spots several millimeters in diameter, most frequently seen in the deep frontal and parietal white matter (Fig. 5.7a), but also seen in the occipital lobes.

Fig. 5.10 Old periventricular leukomalacia. Section of the hemisphere of a child with cerebral palsy, stained for myelin (LBCV). The deep white matter is reduced in volume and pale due to gliosis. Note the thin corpus callosum (arrow) and the dilated square-shaped lateral ventricle.
The earliest stage in the microscopical formation of these lesions is an irregular zone of ‘coagulation necrosis’ in which the tissue loses detailed structure and becomes hyaline and eosinophilic. Nuclei within these zones are pyknotic (Fig. 5.11 and Fig. 5.12). Later, reactive astrocytes and macrophages appear in these infarcted zones. Axons around them show rounded ‘retraction balls’, eosinophilic masses which represent the proximal ends of severed axons. Amyloid precursor protein (APP) is demonstrated in these structures. Mineralization around the periphery of those lesions gives them their chalky appearance (Fig. 5.7a). The central area contains macrophages, which eventually ingest necrotic debris leading to central cavitation.
")
")
Fig. 5.11 Coagulation necrosis. (a) An irregular zone of the white matter stains deeply in an H&E preparation. (x 12.) (b) At higher power it can be seen that most of the nuclei in this zone are pyknotic with condensed, deeply staining chromatin (arrows). (H&E x 450.)
")
")
Fig. 5.12 Periventricular leukomalacia (PVL). (a) Low power of white matter showing early features of PVL. Capillaries are prominent and congested and there is focal hypercellularity. (H&E x 25.) (b) Higher power image shows a reactive capillary with plump endothelial cells (arrow) and increased numbers of glial cells and macrophages in the adjacent white matter. (H&E x 230.)
MULTICYSTIC LEUKOMALACIA

Fig. 5.13 Multicystic leukomalacia. Coronal section of brain. The subcortical white matter has been almost totally replaced by huge cysts. There are smaller cysts in the thalamus, which is shrunken and gliotic (arrow).
The damaged white matter breaks down into multiple cystic cavities, often predominantly subcortical (Fig. 5.13). The cyst walls consist of glial fibers, and white matter is severely reduced in volume. There is extensive neuronal loss in the overlying cortex, the deep gray nuclei and brain stem are also gliotic. Most cases with a documented history seem to occur following abrupt interference in cerebral blood supply between 30 and 44 weeks’ gestation.
POSTERIOR LIMB OF THE INTERNAL CAPSULE
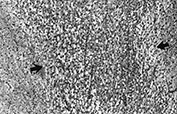
Fig. 5.14 Posterior limb of the internal capsule in an infant whose MRI scan showed marked signal change. The fiber bundle (between arrows) shows intense edema, represented by multiple tiny spaces. (H&E x 45.)
Attention has been drawn to this structure in hypoxic–ischemic damage in neonates by the striking changes identified on MRI scans. In cases where pathology has been correlated with MRI appearances the changes are less striking under the microscope. There is severe edema with little cellular change (Fig. 5.14). Edema in a tightly packed fiber bundle may cause changes of a mechanical nature which are responsible for the MRI appearance9.
< prev | top | contents | next >
Gray matter damage
- Cerebral cortex – mature infants – watershed zones – ulegyria
- Deep gray nuclei – thalamus and lentiform nuclei – status marmoratus
- Brain stem.
CEREBRAL CORTEX

Fig. 5.15 Cortical damage. There is extensive loss of cortex particularly in the depths of sulci: ulegyria (arrows). There has also been loss of white matter with some subcortical cysts. Note the large square lateral ventricle.
The cerebral cortex is damaged by an insult of sufficient severity at any stage but it is characteristically most vulnerable in the term infant. Damage commonly involves the watershed areas: those are the end fields of supply of the major cerebral arteries. The parasagittal cortex is particularly frequently involved. The hippocampus may also suffer.
As in other brain areas the damaged cortex undergoes a stereotyped sequence of cellular reactions. Capillary proliferation in the cortex is often found with abundant capillaries growing in from the adjacent leptomeninges. This accounts for the characteristic appearance of cortical highlighting seen on scans 1 week after injury (Fig. 5.6).
Within the cortex further regional vulnerability is seen. The depths of sulci are particularly likely to be damaged in the mature infant (see Fig. 6.9 and 6.19-6.21). As the lesions evolve the cortex in the depths becomes atrophic while the spared crests of gyri retain their normal thickness (Fig. 5.15). These give a macroscopical appearance likened to ‘mushroom gyri’ and termed ulegyria. This lesion is not described in the immature cortex.
DEEP GRAY NUCLEI
The thalamus and lentiform nuclei are particularly vulnerable to HII. This form of damage occurs at term19 in infants with severe acute insults following, for example, placental abruption. It does occur in very much younger infants4 and may occur antenatally14. The exact site of damage within the basal ganglia and thalami appears to be different in the preterm infant2.
The histological appearances are of cell death, reactive gliosis and capillary proliferation and mineralization. In long-term survivors there is atrophy of these structures, which are densely gliotic. Calcified neuronal profiles may persist (Fig. 5.7b).
STATUS MARMORATUS
Some infants who survive asphyxial damage may show a further peculiar appearance in the deep nuclei. The shrunken nuclei contain irregular bundles of myelinated fibers. This change does not develop at once but only occurs after 6–9 months when active myelination of the deep cerebral structures occurs.
The appearances of status marmoratus on MR imaging have not been described (Fig. 6.20).
BRAIN STEM

Fig. 5.16 Cardiac arrest encephalopathy. There is symmetrical necrosis of the tegmental nuclei in the medulla (arrow), 3 weeks after a severe hypoxic–ischemic episode.
The brain stem nuclei mature very early and are vulnerable to HII. Intrauterine HII to these nuclei is one of the causes of Moebius syndrome.
A striking pattern of brain stem necrosis occurs following acute and total failure of blood supply (cardiac arrest encephalopathy)16. There is symmetrical, bilateral necrosis of the nuclei running throughout the entire length of the brain stem (Fig. 5.16). It is usually associated with lesions elsewhere, particularly in the deep cerebral nuclei.
< prev | top | contents | next >
Malformations (see Chapter 11)
HII is one of the recognized causes of malformation of the brain. For this to occur the insult must occur during the first two trimesters when the brain structures are developing. Malformations of the cortex, such as polymicrogyria, may be seen in watershed territories or on the edges of infarcts arising before 28 weeks22. Insults occurring before 20 weeks interfere with neuronal migration leading to cortical malformation as well as heterotopic neuronal masses in the white matter due to failed migration.
< prev | top | contents | next >
Infections (see Chapter 10)
Viral infections of the developing brain are well recognized. Their effects include neuronal necrosis, gliosis and calcification, which can be very dense and diffuse. Infections in the first two trimesters may cause malformations such as polymicrogryia. The pattern of damage is related to the gestational age at the time of infection as well as the specific agent.
Cytomegalovirus
Cytomegalovirus is the most common viral infection in the developing brain, involving endothelial cells as well as neurons and glial cells. The virus frequently causes polymicrogyria due to its effects on the vascular endothelium15. There is frequently a dense band of calcification in the periventricular tissues.
Herpes simplex
Herpes simplex is rare in utero but may be acquired at birth or in the neonatal period. The effects on the brain are devastating with widespread cystic damage and parenchymal hemorrhage. The appearances on scan may be mistaken for extensive HII following trauma or shaking (Fig. 10.8).
Bacterial infections
Bacterial infections are uncommon in utero, but Listeria is occasionally seen. It causes granulomatous vascular lesions with small focal infarctions (see Fig. 10.17).
Candida

Fig. 5.17 Focal damage due to Candida. There is a rounded mass of cells, reactive glia and macrophages, in the deep white matter. (H&E x 45.)
Candida is the commonest cerebral infection in the neonate. The typical case shows widespread lesions up to half a centimeter in diameter and sometimes cystic, so-called small microabscesses, throughout the cerebral parenchyma, not in a vascular distribution (Fig. 5.17). The organism is readily demonstrated with histological stains as are the presence of microabscesses.
< prev | top | contents | next >
Summary
- Postmortem examination is necessary to define pathological processes in the developing brain.
- An understanding of normal and abnormal brain development is essential to the interpretation of brain scans.
- The brain undergoes a sequence of reactive cellular changes following injury, the nature and severity of which depend on age at insult.
- The patterns of damage show some dependency on age: white matter damage is characteristic of the immature brain while the cortex is vulnerable in the term infant.
- Damage in the first two trimesters may cause malformation.
< prev | top | contents | next >
References
- Back SA, Gan X, Li Y et al. (1998) Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induceddeath caused by glutathione depletion. J Neurosci 18(16), 6241–6253.
- Barkovich AJ and Sargent SK (1995) Profound asphyxia in the premature infant: imaging findings. Am J Neuroradiol 16, 1837–1846.
- Blair E (1996) Obstetric antecedents of cerebral palsy. Fetal and Maternal Medicine Review 8, 199–215.
- Cohen M and Roessman U (1994) In utero brain damage: relationship of gestational age to pathological consequences. Dev Med Child Neurol 36, 263–270.
- Dammann O and Leviton A (1997) Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42(1), 1–8.
- Dammann O and Leviton A (1998) Infection remote from the brain, neonatal white matter damage, and cerebral palsy in the preterm infant. Semin Pediatr Neurol 5(3), 190–201.
- Darrow VC, Alvord EC, Mack LA et al. (1988) Histologic evolution of the reactions to haemorrhage in the premature human infant’s brain. Am J Pathol 130, 44–58.
- Feess-Higgins A and Larroche J-C (1987) Development of Human Foetal Brain: An Anatomical Atlas. Paris, Masson.
- Felderhoff-Mueser U, Rutherford MA, Squier WV et al. (1999) Relation between magnetic resonance images and histopathological findings of the brain in extremely preterm infants. AJNR Am J Neuroradiol 20(7), 1349–1357.
- Gilles FH, Leviton A and Dooling EC (1983) The Developing Human Brain, Growth and Epidemiologic Neuropathology. Boston, John Wright.
- Grafe M (1994) The correlation of prenatal brain damage with placental pathology. J Neuropath Exp Neurol 53, 407–415.
- Hansen A, Leviton A, Paneth N et al. (1998) The correlation between placental pathology and intraventricular hemorrhage in the preterm infant. Pediatr Res 43(1), 15–19.
- Kuban KCK and Gilles FH (1985) Human telancephalic angiogenesis. Ann Neurol 17, 539–548.
- Maalouf E, Battin M, Counsell S et al. (1997) Arthrogryposis multiplex congenita and bilateral midbrain infarction following maternal overdose of coproxamol. Europ J Paed Neurol 5/6, 1–4.
- Marques Dias MJ, Harmant van Rijckevorsel G, Landrieu P et al. (1984) Prenatal cytomegalovirus disease and cerebral microgyria: evidence for perfusion failure, not disturbance of histogenesis, as the major cause of micropolygyria in CMV fetal encephalopathies of circulatory origin. Biol Neonate 50, 61–74.
- Pasternak JF (1993) Hypoxic–ischemic brain damage in the term infant. Pediatr Clin N Am 4, 1061–1072.
- Pryds O and Edwards AD (1996) Cerebral blood flow in the newborn infant. Arch Dis Child 74, F63–F69.
- Roessmann U and Gambetti P (1986) Pathological reaction of astrocytes in perinatal brain injury. Acta Neuropathol (Berl) 70, 302–307.
- Rutherford M, Pennock J, Schwieso J et al. (1996) Hypoxic–ischaemic encephalopathy: early and late magnetic resonance imaging findings in relation to outcome. Arch Dis Child Health 75, F145–F151.
- Squier MV and Keeling JW (1991) The incidence of prenatal brain injury. Neuropathol Appl Neurobiol 17, 29–38.
- Volpe JJ (1989) Intraventricular haemorrhage in the premature infant – current concepts. Part 1. Ann Neurol 25, 3–11.
- Williams RS, Ferrante RJ and Caviness VS (1976) The cellular pathology of microgyria. Acta Neuropathol (Berl) 36, 269–283.
- Yoon BH, Romero R, Kim CJ et al. (1997) High expression of tumor necrosis factor-alpha and interleukin-6 in periventricular leukomalacia. Am J Obstet Gynecol 177(2), 406–411.